This replacement gene can help produce sufficient amounts of the correct protein to to compensate for the deficiency caused by the faulty gene. This is a suitable option when someone has a condition resulting from a mutation in a single, relatively small gene. Viral vectors are generally unable to carry large genes.
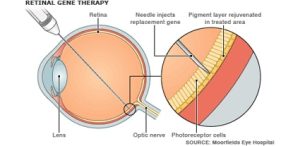
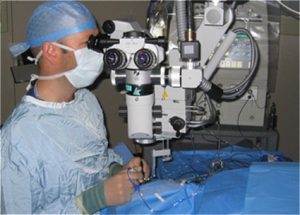
The vector is injected into the back of the eye during surgery. The eye is relatively protected from the body’s powerful immune responses against foreign material like viruses, which might otherwise limit the benefit of gene therapy delivered in this way.
The first gene therapy clinical trial took place in the UK in 2007. Trials have shown encouraging early results, showing that gene therapy can be well-tolerated and improve sight for a number of years. A next wave of studies is now in development to help better understand who might benefit from gene therapy and extend the duration of effect and aspects of sight that can potentially be improved. Find out more about inheritance patterns, genetic testing and genetic counselling.
Gene editing
 Gene editing aims to cut out the damaged part of a gene from a person’s own DNA and replace it with the normal part of the gene. With this technique, scientists can potentially develop effective therapies for much larger genes than current gene therapy techniques allow for and potentially expand the range of conditions that might benefit.
Gene editing aims to cut out the damaged part of a gene from a person’s own DNA and replace it with the normal part of the gene. With this technique, scientists can potentially develop effective therapies for much larger genes than current gene therapy techniques allow for and potentially expand the range of conditions that might benefit.
Several approaches to genome editing have been developed. A recent one, known as CRISPR-Cas9 (or just CRISPR), has generated a lot of excitement in the scientific community because it is faster, cheaper and more accurate than other existing genome editing methods.
Despite developments like CRISPR, gene editing still isn’t very efficient and can result in damage to normal genes, which could cause further harm. Current research is aiming to make this technology more precise so that we can consider using it medically. In the meantime, it is still an extremely useful laboratory technique for investigating diseases in cell and animal models.
Gene therapies will only be of benefit in conditions where retinal cells are still present, just not working correctly. Gene therapy also requires every defective gene to be considered as a different condition requiring its own specific therapy.
Why genes matter
Discover why genes matter and find out more about inheritance patterns, genetic testing and genetic counselling.